Chapter 3 Quick-start
3.1 Installation
3.1.1 Installation from GitHub
This requires the devtools package to be pre-installed first. The HematoMap also required for pre-installation of the Seurat package (https://satijalab.org/seurat/).
# If Seurat is not already installed, you need to install Seurat first
install.packages("Seurat")
# If devtools is not already installed, you need to install devtools first
install.packages("devtools") To install HematoMap
# Installation of HematoMap
devtools::install_github("NRCTM-bioinfo/HematoMap")
# library HematoMap
library(HematoMap)The source code of HematoMap on GitHub can be accessed at https://github.com/NRCTM-bioinfo/HematoMap.
In this chapter, we will describe the main functionalities and a quick-start code template for the workflow of HematoMap. The necessary data for this chapter is already included in the HematoMap package. By reading this chapter, you will learn:
- Gain an overview of the workflow and the built-in database of
HematoMap.
- Gain an overview of the workflow and the built-in database of
- A quick-start code about how to use
HematoMap.
- A quick-start code about how to use
And for the detailed version and advanced application of HematoMap, please read Chapter 4 and Chapter 5.
3.3 Show normal BMMCs
# Show circle plot of normal BMMC
plotCircleTree(group.subc = "reference",
color.mapping = "cell.type",
title = "Normal BMMCs")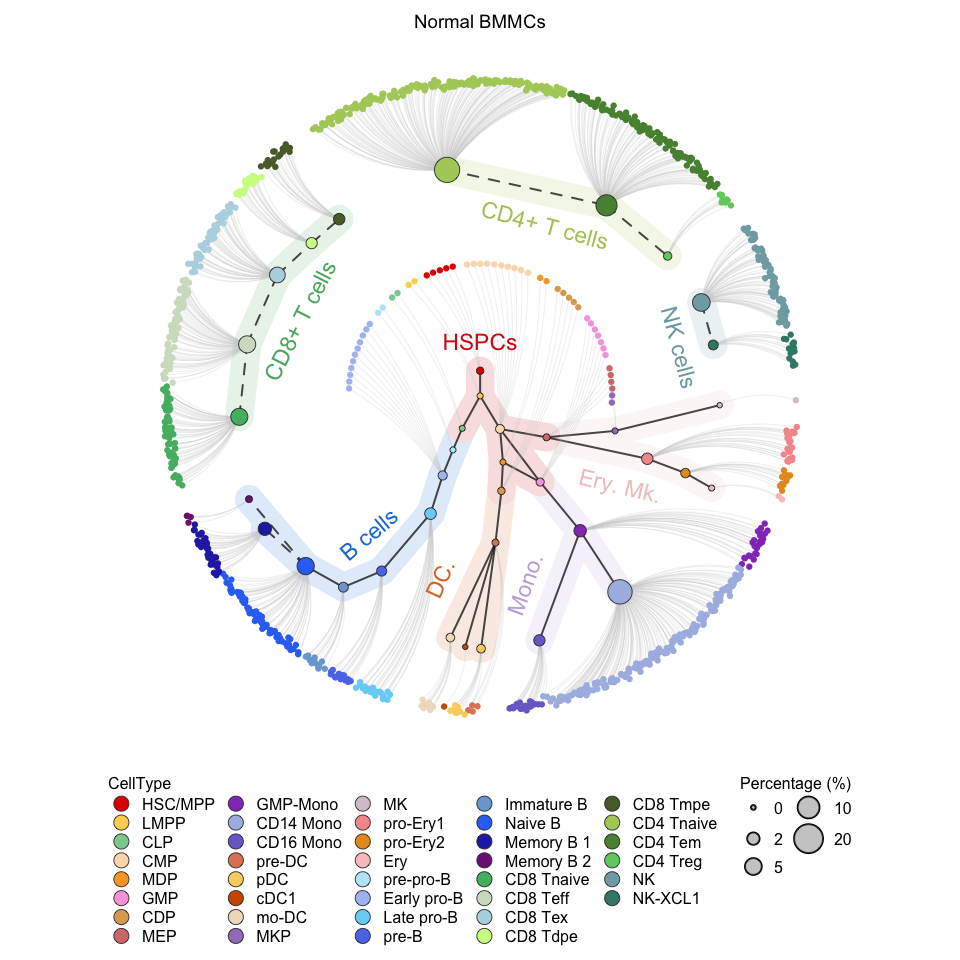
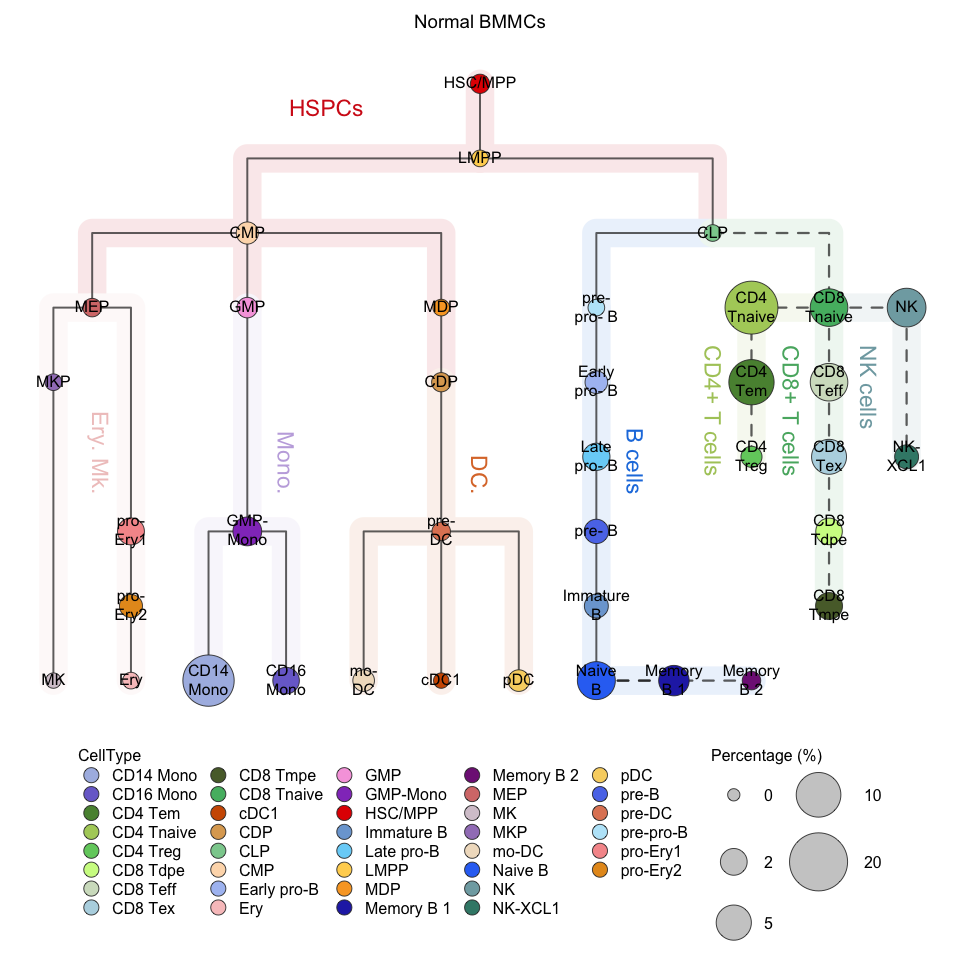
# Show cluster-tree plot of normal BMMC
plotClusterTree(group.subc = "reference",
color.mapping = "cell.type",
title = "Normal BMMCs",
point.size = 20,
label.cell = TRUE)